Rötgenstrukturanalyse – Datenverarbeitung
6. Das Phasenproblem
Das fundamentale Problem der Protein-Kristallografie nennt sich Phasenproblem. Experimentell messen kann man in der Regel nur die Reflex-Intensitäten, aber das ist nur die halbe Wahrheit, da zu jedem gestreuten Röntgenstrahl auch eine Phase gehört, die leider verloren geht. Um eine Elektronendichte (das ist nämlich genau, was gemessen wird, die Verteilung der Elektronenwolken im Kristall) mittels Fourier-Transformation zu berechnen so benötigt man aber beide Informationen: Intensitäten (oder Amplituden) und Phasen. Also muss man die Phasen auf anderem Wege ermitteln, und dafür gibt es 3 grundlegende Methoden:
- Zum einen ist dies der Molekulare Ersatz (molecular replacement). In diesem Falle verfügt man bereits über ein Modell, dass eine grosse Ähnlichkeit mit der untersuchten Struktur aufweist. Die Berechnung der Phasen (inverse Fourier-Transformation) aus dem ähnlichen Modell ist in der Regel ausreichend um die Abweichungen zu modellieren.
- Dann gibt es die direkten Methoden (ab initio phasing). Diese machen sich Relationen zwischen verschiedenen Reflexen zunutze um sich Münchausen artig aus dem Sumpf zu ziehen. Das funktiniert aber leider nicht für grosse Strukturen wie das Ribosom.
- Schliesslich gibt es noch den isomorphen Ersatz (isomorphus replacement, multiple-i. r.), der darauf basiert, dass zwei (oder mehrere) Kristalle gemessen werden, die sich nur durch inkorporation von Schweratomen unterscheiden. Die Schweratome definieren quasi eine Referenzwelle, die als Bezugspunkt für die Phasenberechnung dienen. So reichen dann die Differenzen der Strukturen – also im Prinzip die Substruktur, die nur die Schweratome enthält – aus, um sich die Phasen der ganzen Struktur zu errrechnen.
- Eine Ergänzung oder Alternative ist die anomale Streuung, die darauf basiert, dass an die Nähe einer Absorptionskante eines Atoms, das in der Struktur vorhanden ist, der Streuung eine imaginäre Komponente hinzugefügt wird, so dass die Spiegel-Symmetrie (oder Friedel-Symmetrie) gebrochen wird. Als Folge findet man Differenzen zwischen diesen Spiegel-symmetrischen Reflexen, die genau auf dieselbe Weise verwendet werden können, wie im Falle des isomorphen Ersatzes.

Ta6Br12
Um die Struktur der 30S Untereinheit zu bestimmen mussten wir auf eine Kombination von isormorphem Ersatz und anomaler Streuung zurück greifen, ein homologes Modell war ja nicht vorhanden. Um sicher zu stellen, dass die Differenzen messbar waren (also zumindest grösser als der Messfehler), haben wir grosse, elektronendichte Schweratom-Komplexe wie W18 und Ta6Br12 verwendet.

W18, Cluster mit 18 Wolfram Atomen
Das war ein Glücksfall, denn die W18 – Komplexe haben die Auflösung der Kristalle dramatisch verbessert. Konnten wir bis dahin nur eine Auflösung von 7-9Å erzielen, verbesserte sich durch die Verwendung von W18-Komplexen die Auflösung – und damit die Details, die man erkennen kann – auf bis zu 3Å.
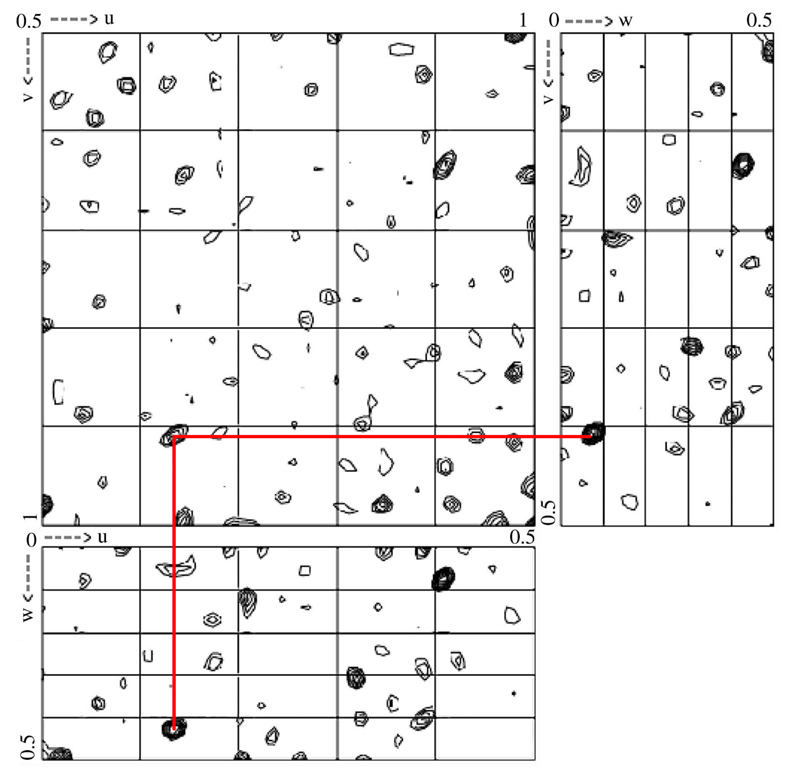
Differenz-Patterson Berechnung, finden sich Maxima in allen drei Diagrammen die zusammenpassen (rot) ergeben sich daraus die Koordinaten für eine Schweratomposition.
Dummerweise waren damit die Kristalle mit und ohne W18 nicht mehr isomporph, die Differenzen damit praktisch nutzlos. Legt aber die Kristalle mit W18-Komplexen für kurze Zeit in eine Kristallösung ohne W18 Zugabe, so lassen sich einige der W18-Komplexe wieder aus den Kristallen ‚heraus waschen‘, so dass wir die Differenzen doch noch verwenden konnten um die Positionen der W18-Komplexe im Kristall zu ermitteln (z.B. im Differenz-Patterson rechts), und damit die Phasenberechnung beginnen konnten.
Obwohl man eigentlich angenommen hatte, dass die grossen Komplexe nur zur Phasierung bei niedriger Auflösung taugen (irgendwann kommen destruktive Interferenzen zum Tragen, die die Differenzen zu klein werden lassen um genaue Phasen damit zu ermitteln), konnten wir die Struktur bis zu einer Auflösung von 3Å bestimmen, allein basierend auf den isomorphen und anomalen Differenzen der W18-Komplexe, ein hartes Stück Arbeit und viele kurze Nächte.
 Dennoch braucht man aber deswegen nicht gleich Physik zu studieren, denn zum Glück werden alle diese Berechnungen heutzutage von verschiedenen Programmen durchgeführt, deutlich schneller als noch vor 5 Jahren. In vielen Fällen braucht man nicht einmal diese Programme studieren um zum ersten Strukturentwurf zu kommen.
Dennoch braucht man aber deswegen nicht gleich Physik zu studieren, denn zum Glück werden alle diese Berechnungen heutzutage von verschiedenen Programmen durchgeführt, deutlich schneller als noch vor 5 Jahren. In vielen Fällen braucht man nicht einmal diese Programme studieren um zum ersten Strukturentwurf zu kommen.
Leider ist es bei Ribosomenkristallen selten so einfach. Nach der Schwerstarbeit der ersten Strukturen konnte man neuere Komplexe dieser Strukturen aber nach der Methode des Molekularen Ersatzes (siehe 1.) berechnen.

W18-Cluster bindung an einem Protein. Hier stabilisiert der Cluster die sonst flexiblen Enden und einen Loop.

Einige W18 am 30S.
Interessanterweise binden die Wolfram-Cluster (W18) fast ausschließlich an den ribosomalen Proteinen (nur eine mögliche Bindunsstelle an der RNA konnten wir finden).
Dabei werden oftmals die flexiblen ‚Arme‘ verschiedener Proteine zur Bindung mit einbezogen (bei S2, S11, S18…). Das wiederum liegt den Schluß nahe, daß diese langen, unstrukturierten Arme bei der Bindung von Faktoren (IF1-3) oder tRNA, mRNA oder großen Untereinheit eine wichtige Rolle spielen.
In unserem Fall hat das aber vor allem zur Stabilisierung der Struktur und damit der Verbesserung der Auflösung geführt – siehe oben …
Nachtrag 2012:
Mittlerweile gibt es etliche Proteinkristallographie-Beamlines mit ‚Full-Service‘, d.h. man liefert die Kristalle und bekommt die Strukturen. Der ganze kristallografische Teil der Strukturlösung wird von den Betreueren oder lokalen Mitarbeitern der Messplätze übernommen.
Nachtrag 2015:
Die aktuellste Weiterentwicklung ist die Messung von Mikrokristallen oder sogar Einzel-Molekülen an Freie-Elektronen-Lasern (z.B. LCLS in Stanford/USA, XFEL demnächst in Hamburg). Bei dieser Methode werden mit ultrakurzen Röntgenblitzen Diffraktionsaufnahmen von einzelnen Mikrokristallen oder sogar nur einezelnen Molekülen gemacht und aus millionen dieser Aufnahmen ein Datensatz zur Strukturbestimmung zusammengestellt. Meist hilft dabei aber auch noch der Molekulare Ersatz (siehe 1.).
7. Elektronendichte, Modellierung
. Auch der Laie kann erkennen, daß die Modellierung der linken Dichte garantiert leichter fällt alls bei den beiden Anderen.")
Ein Beispiel dafür, wie wichtig die richtige Wahl der Parameter bei der Phasierung ist (Bild von Dr. Marco Glühmann). Auch der Laie kann erkennen, daß die Modellierung der linken Dichte garantiert leichter fällt alls bei den beiden Anderen.
Hat man letztendlich die Strukturfaktor-Beträge und eine möglist gute Annäherung an deren reale Phasen, kann man beginnen ein Modell der atomaren Struktur in die berechnete Elektronendichte einzupassen.
Es gibt mittlerweile auch gut funktionierende Programme die anhand der Aminosäure-Sequenz der Proteine, diese automatisch in die Elektronendichte einpassen. Allerdings hängen die Erfolgsaussichten stark von der Qualität der Dichte, der Auflösung und der Größe der zu modellierenden Struktur ab.
Ribosomale Multi-Protein-RNA Riesenmoleküle lassen sich bisher leider nur in ‚Handarbeit‘ lösen. Zuerst einmal werden alle bekannten Informationen zusammengesucht, die auch nur irgendeine Aussage über die Struktur treffen z.B.:
- Crosslink Daten, d.h. welche Proteine liegen dicht beieinander oder welchen Teilen der RNA kommen sie nahe usw.
- Biochemische Information über bestimmte Funktionen des ribosomalen Partikels und beteiligten Substrukturen.
- Infomationen über homologe Strukturen, die bereits als atomare Modelle vorliegen wie z.B.:
- Strukturen ribosomaler Proteine in ungebundener Konformation
- typische RNA-Motive
- Kontaktbereiche zur anderen Untereinheit
- Zwei- oder dreidimensionale Vorhersagen der Proteinstruktur
Dann kommt es auf die Qualität und Auflösung der Elektronendichte an. Hat man eine Auflösung von 3A oder höher, in guter Qualität, dann kann man relativ einfach RNA- und Protein-Dichte unterscheiden. Man kann mit den RNA-Helix Bereichen anfangen, die Länge der modellierten Helices bestimmen, eventuell die mono-Strang RNA Verbinungen und Enden (Loops) modellieren soweit dies möglich ist. Parallel dazu versuchen die einzelnen Teile der zweidimensionalen Strukturvorhersage der RNA zuzuordnen.
Da mit Sicherheit nicht nur eine Person an dem Projekt arbeitet kann man parallel zur RNA die Protein-Dichte modellieren, den Anfang bilden auch hier die größeren Teile, also alpha-Helices. Dann vielleicht beta-Stränge oder die beta-’sheets‘, Kombinationen von mehreren beta-Strängen. Hat man dieses, lassen sich schon anhand der strukturellen Bestandteile viele Proteine den einzelnen Dichtebereichen zuordnen. Mit Wissen der Sequenz oder noch einfacher, mit einer homologen, bekannten Struktur lassen sich dann die fehlenden Teile noch einfacher Modellieren. Irgendwann ist dann die Struktur soweit gediehn, dass man mehr möchte als das große Puzzle beenden.
. Rechts: Gutre Qualität der hohen Auflösungsdaten, mit Hilfe des umgebenden Modells zusätzlich Phasierungshilfe durch molekularen Ersatz.")
Links: Wenig Informationen, niedrige Auflösung oder qualitativ schlecht im Bereich der hohen Auflösung (7-3A). Rechts: Gute Qualität der hohen Auflösungsdaten, mit Hilfe des umgebenden Modells zusätzlich Phasierungshilfe durch molekularen Ersatz.
Ist man in der unglücklichen Situation eine weniger starke Auflösung und Elektronendichte von geringerer Qualität zu haben (wie es bei unserem 30S anfangs der Fall war), dann ist die Puzzlearbeit weitaus schwieriger.
 der atomaren Struktur. Viele Details, z.B. einzelne Basen sind noch nicht zu sehen.")
Eine RNA-Helix: Das Strukturmodell (gelb) eingepasst in eine frühe Elektronendichte. Mit höherer Auflösung und bessere Qualität lassen sich dann die einzelnen Basenpaare in der Dichte erkennen.
Sicherlich fingen auch wir mit den typischen RNA-Helix Teilen an, und auch die alpha-Helizes einiger Proteine waren schnell zu modellieren. Dann aber gerät man ein wenig ins Stocken, denn auch wenn das Teilmodell mit zur Verbesserung der Elektronendichte beiträgt, wesentliche Bestandteile waren oft mehr als unklar. Viele Verbindungen zwischen Helizes werden mehr geraten als modelliert, vor allem Kombinationsgabe und Puzzlewütigkeit ist dann gefragt. Alle externe Information mit dem Teilmodell kombinieren und versuchen Schritt für Schritt die Teile zuzuordnen und zu verbinden.

Beispiel für einen Protein-Loop. Bei ca. 3A lassen sich bereits die meisten Seitenketten der Aminosäuren erkennen und teilweise auch modellieren.
Das Gefühl, nach tage- oder wochenlanger Grübelei endlich die Lösung zu haben ist unbeschreiblich (‚da geht mir ein Licht auf‘).
Das ‚finetuning‘ ist bei dieser Auflösung ungleich schwieriger, sind doch die Details der Proteinstruktur nur schwerlich oder gar nicht zu erkennen. Meist gibt man sich erstmal mit der C-alpha-Kette, dem sog. Rückrat der Aminosäurekette zufrieden. Die wenigen Details (Seitenketten) nutzen lediglich zur Modellierung der richtigen Sequenz.
8. Am Ziel, die Struktur
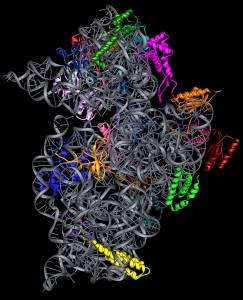 Am Ende hat man die faszinierende Struktur vor sich. Nach all den Jahren (für Ada Yonath waren es fast 20 seit den ersten Kristallen). Vielleicht ist das Ergebnis noch mit ein paar kleinen Fehlern behaftet (je nach Auflösung), aber garantiert ist eines: Die Struktur ist SCHÖN.
Am Ende hat man die faszinierende Struktur vor sich. Nach all den Jahren (für Ada Yonath waren es fast 20 seit den ersten Kristallen). Vielleicht ist das Ergebnis noch mit ein paar kleinen Fehlern behaftet (je nach Auflösung), aber garantiert ist eines: Die Struktur ist SCHÖN.
Nun muß man nur noch schneller publizieren als die Konkurrenz (falls es welche gibt).
Ist die Hauptarbeit getan, treten oftmals die Trittbrettfahrer auf den Plan und versuchen die größten Stücke vom Kuchen abzubekommen, und so ist die eigentliche Schönheit der Struktur nur von kurzer Dauer (Wissenschaft ist leider nicht nur Spaß sondern oftmals auch reine Politik).
Und dann ?
Hat man die Struktur geht die Arbeit weiter, man kann über Funktion(-en) sinnieren, versuchen Faktoren oder Antibiotika zu binden. …. Dann gibt es ja noch weitere Untereinheiten oder einfach eine andere Bakterienart und und und …. das kann so weiter gehen bis einem der Geldhahn abgedreht wird, der/die Chef/in in Rente geht, einem die Ideen ausgehen, der Nobelpreis vorbeischaut (obwohl das einen höchstens nur zeitlich hindern sollte weiterzumachen) oder das bis Herz einen wieder –> ’nach Hause‘ treibt …